Trait evolution on phylogenetic networks
We assume a fixed network, correctly rooted, with branch lengths proportional to calendar time. Note that SNaQ will estimate branches in coalescent units, and we need to calibrate the network so that branches are proportional to calendar time. We will skip this step here and provide the calibrated network, but you can do this calibration in BPP or BEAST.
We want to be inside the analysis folder.
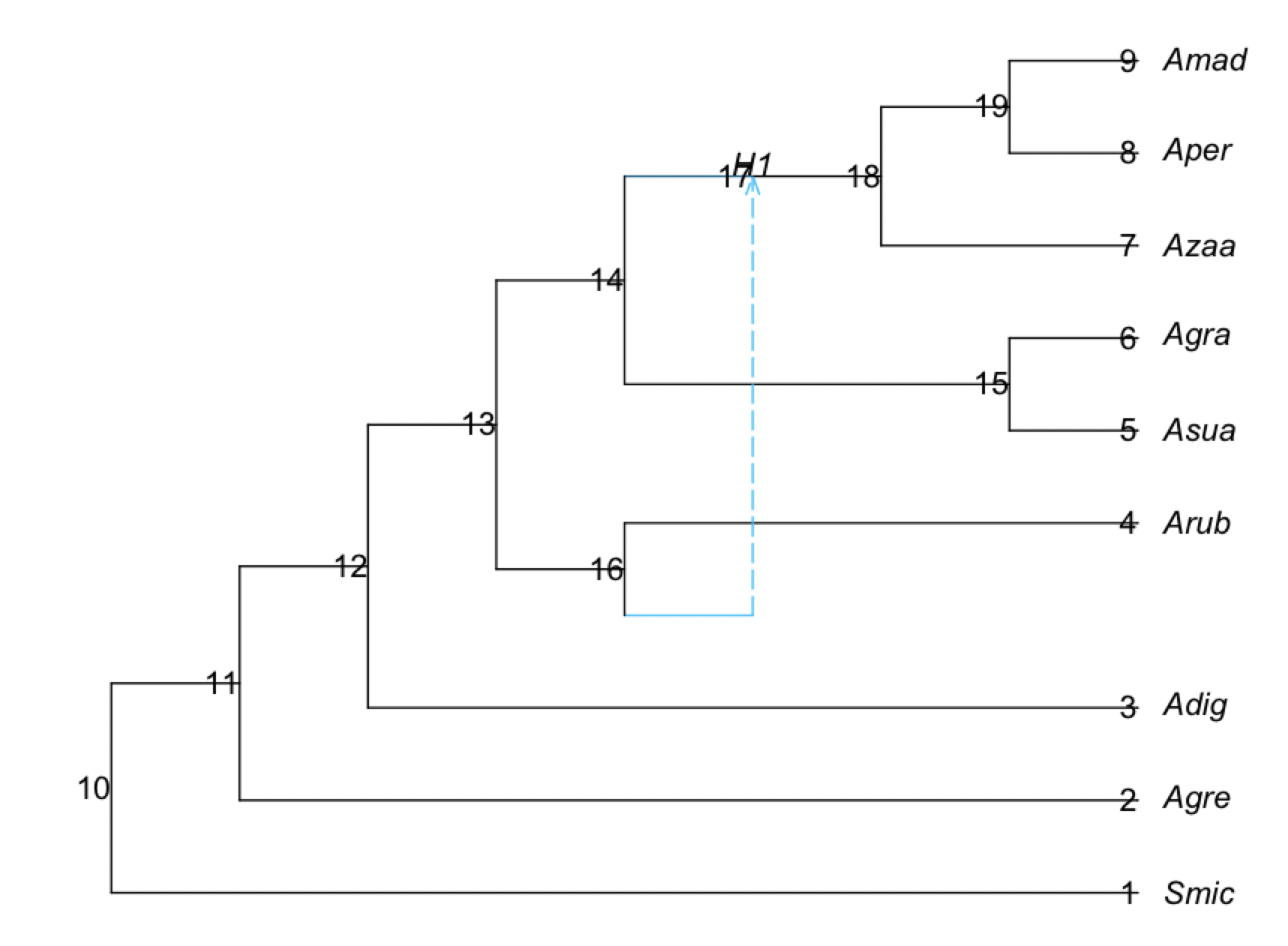
net1 = readnewick("(Smic:18.12505298,(Agre:8.747492442,(Adig:8.308140028,((#H1:0.0::0.117935,Arub:3.476676179):2.658717826,((Asua:3.171531094,Agra:3.171531094):2.767496398,((Azaa:1.381629136,(Aper:1.363263922,Amad:1.363263922):0.01836521346):2.095047043)#H1:2.460938604::0.882065):0.1963665132):2.172746023):0.4393524143):9.377560535);")
rootatnode!(net1, "Smic")
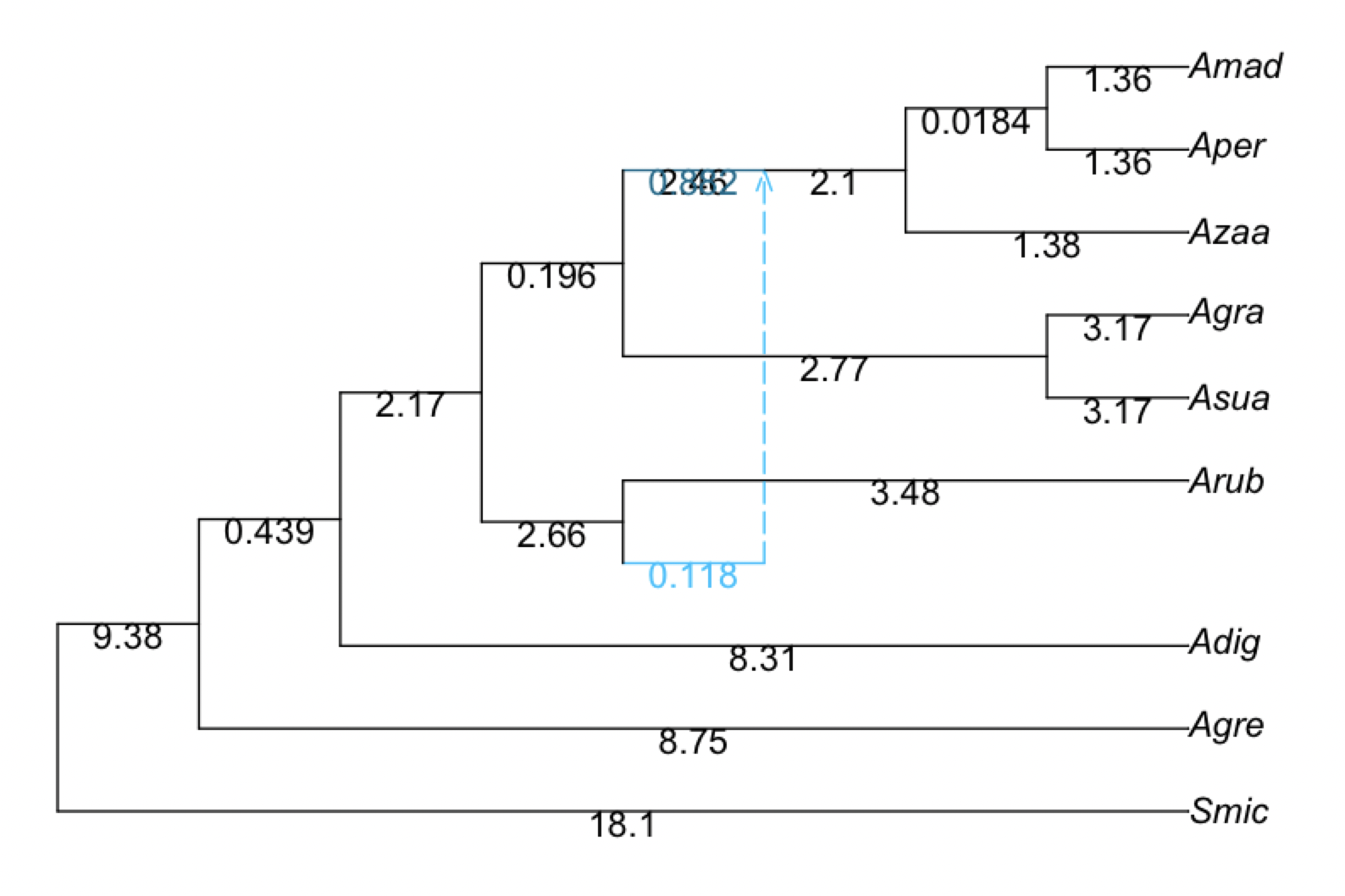
plot(net1, showgamma=true, showedgelength=true)
Now, we want to read the trait data (denoted traitData.txt in the data folder).
In the shell:
$ less ../data/traitData.txt
Species Short_Name Flower_Color Pollination_Syndrom
A_digitata Adig white mammal
A_suarezensis Asua white mammal
A_grandidieri Agra white mammal
A_za Azaa pigmented hawkmoth
A_madagascariensis Amad pigmented hawkmoth
A_perrieri Aper pigmented hawkmoth
A_rubrostipa Arub pigmented hawkmoth
A_gregorii Agre white hawkmoth
S_micrantha Smic white both
In julia:
using CSV, DataFrames
df = CSV.read("../data/traitData.txt", DataFrame, stringtype=String)
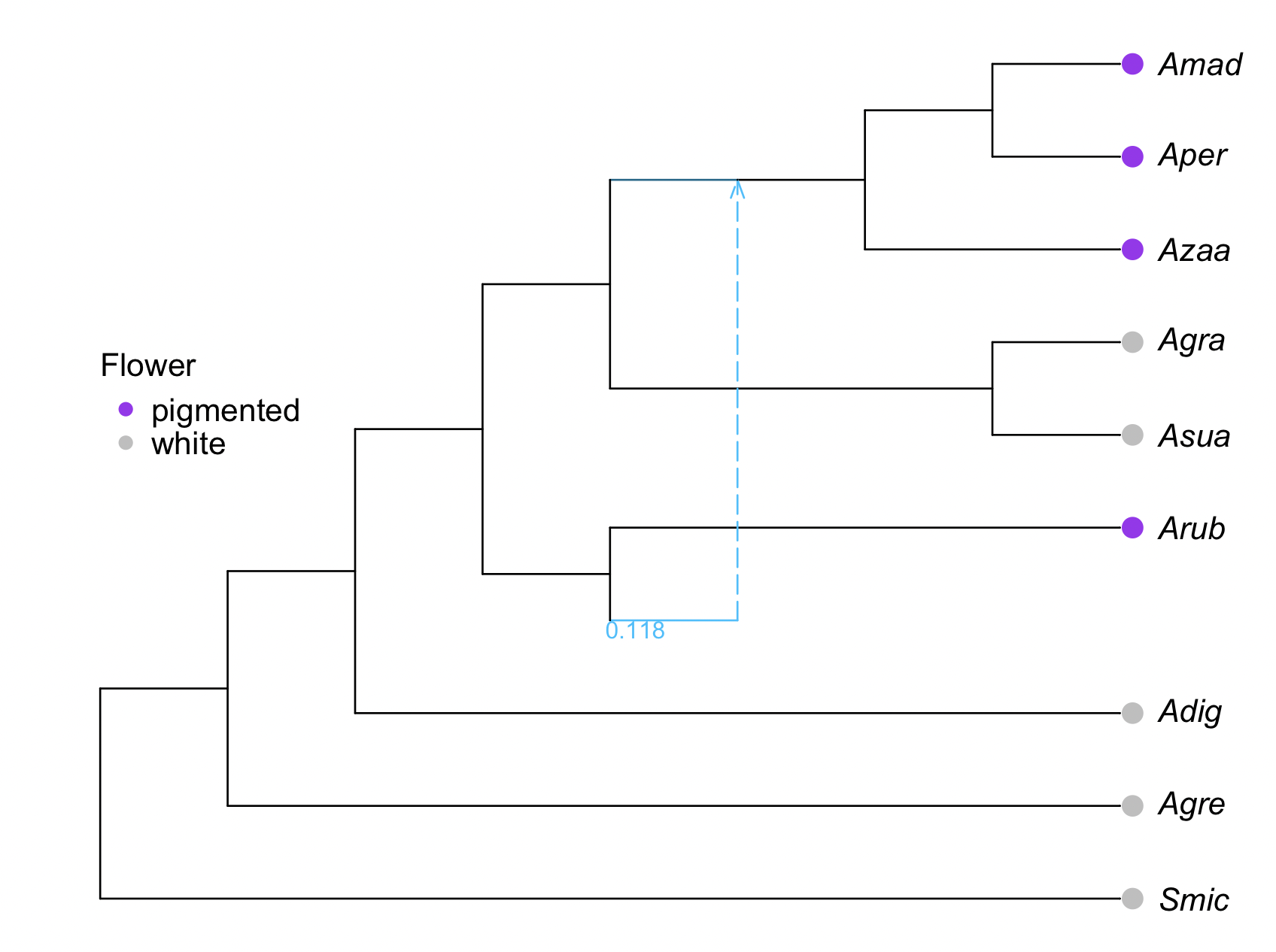
First, we will try to plot the traits. We need to remove the species columns from the data:
species = df.Short_Name # or: dat[!, :species]
select!(df, Not(:Short_Name)) # select all columns except for :Short_Name; modifies dat in place
select!(df, Not(:Species))
select!(df, Not(:Pollination_Syndrom))
using RCall, PhyloPlots
R"par"(mar=[0,0,0,0]); # to reduce margins
res = plot(net1, tipoffset=0.3); # the results "res" provides point coordinates, to use for data annotation
o = [findfirst(isequal(tax), species) for tax in tipLabels(net1)] # 5,2,4,1,3,6: order to match taxa from "species" to tip labels
isequal(species[o], tipLabels(net1)) # true :)
traitcolor = map(x -> (x=="white" ? "grey" : "purple"), df.Flower_Color[o])
leaves = res[13][!,:lea]
R"points"(x=res[13][leaves,:x] .+0.1, y=res[13][leaves,:y], pch=16, col=traitcolor, cex=1.5); # adds grey & red points
R"legend"(x=1, y=7, legend=["pigmented","white"], pch=16, col=["purple","grey"],
title="Flower", bty="n",var"title.adj"=0);
# next: add to gene flow edge the proportion γ of genes affected
hi = findfirst([!e.ismajor for e in net1.edge]) # 6 : "h"ybrid "i"ndex: index of gene flow edge (minor hybrid) in net: horizontal segment
R"text"(res[14][hi,:x]-0.3, res[14][hi,:y]-0.1, res[14][hi,:gam], col="deepskyblue", cex=0.75); # add the γ value
Inference of evolutionary rates
Choose a model to describe how evolutionary changes happened over time. For general trait types, use one of these three Markov substitution models:
:BTSMBinary Trait Substitution Model (2 states, rates unconstrained):ERSMEqual Rates Substitution Model (k states, all transitions possible with equal rates):TBTSMTwo Binary Trait Substitution Model (though not fully implemented yet)
To infer evolutionary rates, run fitdiscrete on the network and data. It will calculate the maximum likelihood score of one or more discrete trait characters at the tips on a fixed network with the following assumptions:
- Along each edge, evolutionary changes are modeled with a continous time Markov model.
- At a hybrid node, the trait is assumed to be inherited from one or the other of its parents (immediately before the reticulation event), with probabilities equal to the inheritance γ of each parent edge, which is given by the network.
- At the root of the network, a uniform distribution among the possible states is assumed a priori.
- The model ignores incomplete lineage sorting (e.g. hemiplasy).
using PhyloTraits
s1 = fitdiscrete(net1, :ERSM, species, df; optimizeQ=false)
where the arguments are:
net1is the estimated (calibrated) network:ERSMis the substitution modelspeciesis an array with the species names at the tipsdfis the dataframe that has the trait (Flower color in this case)optimizeQ=falsemeans that the transition rates will stay at their starting values, without being optimized.
PhyloNetworks.StatisticalSubstitutionModel:
Equal Rates Substitution Model with k=2,
all rates equal to α=0.01403.
rate matrix Q:
white pigmented
white * 0.0140
pigmented 0.0140 *
on a network with 1 reticulations
data:
9 species
1 trait
log-likelihood: -6.10081
Ancestral state reconstruction
The command to reconstruct the ancestral states is:
ancestralreconstruction(s1)
which provides the following output:
julia> ancestralreconstruction(s1)
19×4 DataFrame
Row │ nodenumber nodelabel white pigmented
│ Int64 String Float64 Float64
─────┼───────────────────────────────────────────────
1 │ 1 Smic 1.0 0.0
2 │ 2 Agre 1.0 0.0
3 │ 3 Adig 1.0 0.0
4 │ 4 Arub 0.0 1.0
5 │ 5 Asua 1.0 0.0
6 │ 6 Agra 1.0 0.0
7 │ 7 Azaa 0.0 1.0
8 │ 8 Aper 0.0 1.0
9 │ 9 Amad 0.0 1.0
10 │ 10 10 0.956058 0.0439417
11 │ 11 11 0.980099 0.0199013
12 │ 12 12 0.977674 0.0223259
13 │ 13 13 0.86375 0.13625
14 │ 14 14 0.864546 0.135454
15 │ 15 15 0.993368 0.00663154
16 │ 16 16 0.264048 0.735952
17 │ 17 H1 0.217158 0.782842
18 │ 18 18 8.94943e-5 0.999911
19 │ 19 19 5.25928e-5 0.999947
The white and pigmented columns correspond to the probability that the state in a given node correspond to each of these trait values. The rows correspond to different nodes, with the internal nodes represented by their node number.
To know to which node each row correspond, we can plot the network with node numbers:
plot(s1.net, shownodenumber=true, shownodelabel=true, tipoffset=0.2);